技术性贸易措施公共服务平台
印度防护用品市场准入介绍
2020-03-31 14:02(一)印度医疗器械市场概述
在印度,医疗器械包括有助于诊断和治疗的特定器械、影响人体的特定物质、外科手术服、绷带、采血袋以及用于诊断或治疗任何人或动物的任何疾病或紊乱的其他物质或器械。具体的设备和物质将由政府不定时通知。医用口罩、手套、防护服等皆属于医用&医疗器械产品。
印度医用&医疗器械设备市场跻身全球前20名,是亚洲第四大市场。其市值约为55亿美元,预期年复合增长率为15%。在过去的二十年里,印度市场经历了不断的变化。在1991年新经济政策出台之前,印度国内制造业处于产业主导地位。但之后它变成了一个进口驱动的市场。
(二)医疗器械技术法规
在“印度制造”计划的支持下,2017年1月31日,卫生和家庭福利部( Ministry of Health & Family Welfare)颁布了《医疗器械条例,2017》,自2018年1月1日起生效,并于2018年6月1日颁布了《医疗器械条例补充条款,2018》。在2017年《医疗器械条例》实施之前,印度根据《药品和化妆品法,1945》将医疗器械作为药物(制药产品)进行监管,因此,该条例规定医疗器械应与药物分开。
新规符合全球协调工作组(GHTF)框架,符合最佳国际惯例,并根据风险对医疗器械进行分类。根据新规,医疗器械被分类为A级(低风险)、B级(低至中度风险)、C级(中至高风险)和D级(高风险)。2017年11月1日,药品管理局(Drugs Controller General,DCGI)根据《医疗器械条例,2017》的规定,通报了351种医疗器械和247种体外诊断医疗器械及其风险等级。这份清单详细列出了之前根据《药品和化妆品法》通报的15类医疗器械和8种物质。
新规的制定是为了促进印度国内制造业的发展,并规范该地区的进口和制造业。此外,新的医疗器械规则中也引入了公告机构的检查。《医疗器械条例,2017》介绍了由公告机构进行的第三方合格评定。印度认证机构国家认可委员会(NABCB)已被被中央许可证颁发机构(CLA)确定为授权认可机构。公告机构将对A类和B类医疗器械制造商的质量管理体系进行核查和评估,如有需要,还将被要求协助对C类和D类医疗器械进行监管。制造商的质量管理体系必须符合ISO 13485。
(三)医用及医疗器械规管机构
中央药品标准控制机构(Central Drugs Standard Control Organization,CDSCO),是印度卫生与福利部(Ministry of Health and Family Welfare)下属的监管医疗器械进口、生产和销售的重要部门。中央药品标准控制机构曾经是管理药品、诊断试剂和化妆品的标准、法规、生产和进口的部门。现在,CDSCO的职能是制定药品、疫苗、血液制品和注射液的标准、法规。同时该机构增加了监管医疗器械的职能,为了加强对医疗器械的监督管理。
印度认证机构国家认可委员会(NABCB)由印度工商部下的印度质量委员会建立的,其目的是作为国家认可机构认证公告机构。国家认可委员会确定公告机构的合格评定活动及认可标准,制订公告机构的认可准则及程序,并定期对公告机构进行审核,以评估其是否符合医疗器械规则。
(四)医用&医疗器械产品标准
印度医疗器械产品遵循的标准总结如下:
-制造商必须遵循中央政府为医疗器械专门制定的或BIS制定的标准;
-若未制定任何医疗器械的相关标准,则该器械应符合国际标准化组织(ISO)或国际电工委员会(IEC)或任何其他药典标准制定的标准;
-如果没有上述标准类别,则应采用经验证的制造商标准即客户标准。
印度标准局BIS针对包括基础和生产工程、化工、城建、电子和通信、食品与农业、机械工程、管理和体系、医药设备和医院规划等15个领域设立了相应的16个分部理事会,将标准制定工作分配给涉及不同的技术范围的分部理事会。其中,口罩、手套以及防护服等相关标准主要涉及医药设备和医院规划(Medical Equipment and Hospital Planning Department,MHD)分部委员会,化工(Chemical Department ,CHD)以及纺织(Textile Department ,TXD)分部委员会也制定了少量标准。
表1: 印度医用防护产品标准
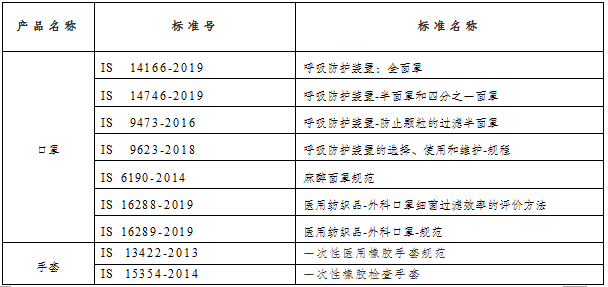
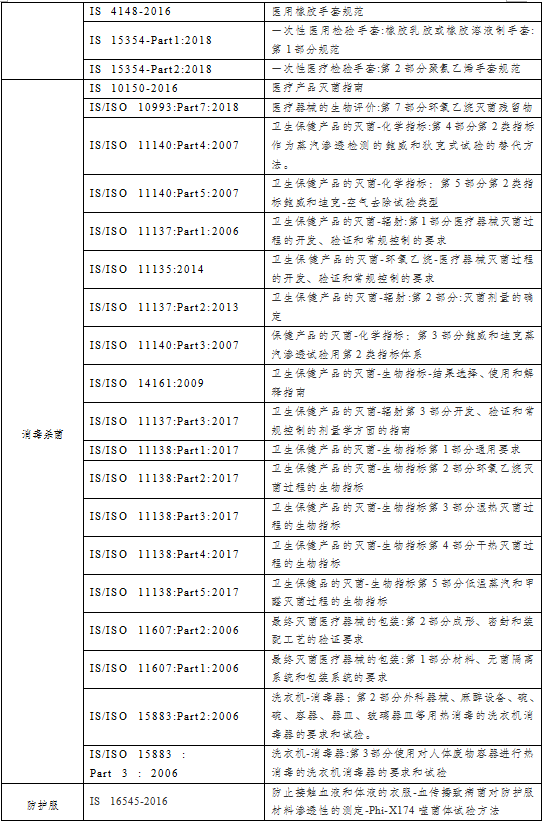
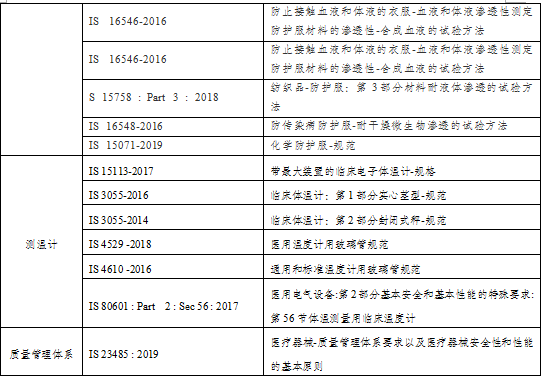
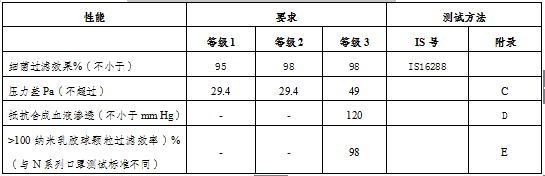
表2:印度IS 16289医用纺织品外科口罩规范
(五)医疗器械的进口
在印度,注册证书和进口许可证是印度医疗器械制造商进口任何医疗器械的强制性要求。如果一家医疗器械企业想进入印度市场,但在印度没有注册办事处,则需要CDSCO授权的印度代理才能这样做。
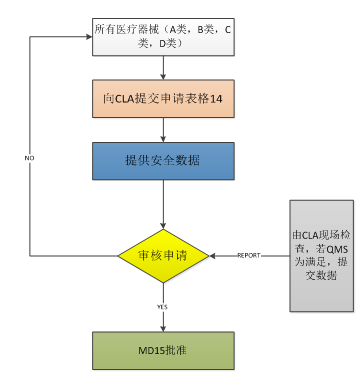
图1:医疗器械进口许可证审批
所有医疗器械产品进口都需要获得印度药品管制局ADC 的无异议证书(No Objection Certificate,NOC)。协助药品管制员(ADC)会询问药品的进口许可证。如进口原材料,那么协助药品管制员(ADC)会要医疗器械产品的生产许可证。其他进口清关重要还有商业发票、装箱单、货运证书、保险证书、原产地证书以及空运单/提货单。
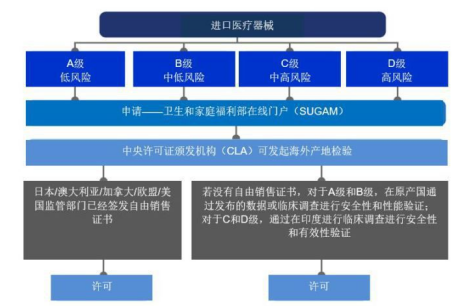
图2:医疗器械进口流程
(六)医疗器械产品上市
目前,印度A类产品不需要许可证,但其自愿申请须由州许可当局(SLA)许可;B类设备需要(SLA)审批;C类和D类设备需要中央许可局(CLA)审批。
1、非管制类型:
目前,印度只有40-50个医疗器械需要注册。对于所有其他不需要注册的医疗器械,制造商应从DCGI获得无异议证书(NOC)。NOC是来自DCGI的一封信,声明该产品不需要注册,可以自由进口到印度。
2、管制类型:
医疗器械要在印度上市,需要申请医疗器械注册证,注册证书有效期为三年,需要在证书有效期九个月前提交重新注册申请。医疗器械注册主要分为四步:
(1)支付费用。
(2)准备资料。印度进口医疗器械注册主要参考文件为《印度医疗器械注册指南》.该指南中详细介绍了医疗器械注册需要准备的文件,以及文件中需要包括的具体内容。其中企业主文档和产品主文档为其中涵盖内容最多也是最重要的两份文件。需要准备文件如下:
a. 附信。随提交文件一起提交的说明信。这封说明信非常重要,信中应详细说明产品的注册是首次注册还是重新注册,还是扩增产品注册。
b. 申请表。申请表中包括制造商信息,申请产品信息,以及需要交纳费用等信息。
c. 原始收据。缴纳注册相关费用的原始收据。
d. 授权信。制造商授权印度代理商的一份证明性文件,内容包括双方的公司名称地址,要注册产品的名称型号等信息。应注意,授权信需要在制造商所在地国家的印度使馆进行认证。
e. 自由销售证明书。原产地的自由销售证明书或在美国、加拿大、日本、澳大利亚和欧盟的自由销售证明书(如适用)。原产地的自由销售证明书应在制造商所在地国家的印度使馆进行认证。
f. 依据ISO13485颁发的体系证书。
g. C E证书。应在制造商所在地国家的印度使馆进行认证。
h. CE设计确认证书(如适用)。只要在欧盟分类为第三类的产品才有CE设计确认证书, 也应在制造商所在地国家的印度使馆进行认证。
i. 符合性声明。
j. 审核报告。
k. 企业主文档。从企业概况,生产产品的介绍到企业机构与人员、厂房和设施、设备、生产、质量控制、储存、文件、抱怨和纠正预防措施等各个方面的要求,全面详细地介绍体系文件。虽然文件与ISO 13485非常相似,但是要按照附录V的要求整理一份企业主文档,是一件费时费力的事情。同时,在《印度医疗器械进口注册问题汇总》中47条说到,按照ISO 13485制定的《质量手册》,在医疗器械注册中可提交质量手册来替代按照指南附录V准备的企业主文档。
l. 产品主文档。按照指南的附录VI编写产品主文档。包括内容基本与CE认证技术文档一致,但是要按照附录VI的要求编写,主要内容有:
①产品综述:简要介绍产品预期用途;结构;有效期;是否无菌;价格;在其他国家的注册情况;不良事件以及整改措施等。
②详细的产品描述以及规格介绍:产品预期用途;禁忌症;材质;适用人群;结构说明;附件的说明;主要功能部件的说明;规格型号的详细区别及描述;以及对比产品的情况说明。
③包装标签:主要包括产品的宣传册,操作手册,以及产品的包装(应包括产品所有附件的包装以及标签)。按照印度《药品和化妆品法》,注册时要提供一整套产品的包装原件(应包括产品所有附件的包装以及标签)。
④设计和生产信息:设计资料包括产品生产流程图和产品设计确认资料。生产信息包括产品生产概况;生产环境;生产设备;产品组装;半成品、成品检验;产品的包装及储存信息。如果产品的生产在不同的生产地址,应对不同生产地址进行有效的区别。
⑤适用基本标准列表:该产品适用哪些标准,以及相关的检测报告或符合性说明。
⑥风险分析报告。
⑦产品确认和验证报告:包括工艺验证报告;生物安全性试验;无菌验证报告;软件验证确认报告;动物试验;加速老化试验;临床资料;上市后的检测数据。
3、提交产品注册申请到CDSCO。
4、获证。如果提交资料齐全,CDSCO会在9个月之内颁发医疗器械注册证书。
(七)进口医疗器械上市后监管
医疗器械上市之后,一旦出现不良事件,制造商必须向CDSCO报告。当医疗器械被强制撤出其他国家市场,或是取消合法许可时,也必须撤出印度市场,这种撤出是暂时性的,然后CDSCO会进一步讨论合理的管理活动。制造商在申请注册时即被要求进行上市后监督管理,包括产品分销记录程序、投诉处理、不良事件报告和产品召回程序等。2
参考资料:
1 TUV南德认证检测
2秦韶燕, 崔涛, 殷海松. 印度对医疗器械市场准入的要求[J].《中国医疗器械杂志》2016年01期。
![]()